
1.3: The 1st and 3rd laws of thermodynamics, and heat capacity




- Last updated
- Sep 20, 2022
- Save as PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
Now let us consider a thermally insulated system whose volume V may be changed by force – see, for example, Figure 1.1.1. Such a system is different from the fully closed one, because its energy E may be changed by the external force’s work – see Equation (1.1.1):
dE=dW=−PdV.
Let the volume change be so slow (dV/dt→0) that the system is virtually at equilibrium at any instant. Such a slow process is called reversible, and in the particular case of a thermally insulated system, it is also called adiabatic. If the pressure P (or any generalized external force Fj) is deterministic, i.e. is a predetermined function of time, independent of the state of the system under analysis, it may be considered as coming from a fully ordered system, i.e. the one having zero entropy, with the total system (the system under our analysis plus the source of the force) completely closed. Since the entropy of the total closed system should stay constant (see the second of Eqs. (1.2.2) above), S of the system under analysis should stay constant on its own. Thus we arrive at a very important conclusion: at an adiabatic process, the entropy of a system cannot change. (Sometimes such a process is called isentropic.) This means that we may use Equation (???) to write
P=−(∂E∂V)S.
Now let us consider a more general thermodynamic system that may also exchange thermal energy (“heat”) with its environment (Figure 1.3.1).
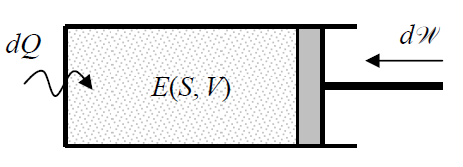
For such a system, our previous conclusion about the entropy’s constancy is not valid, so that S, in equilibrium, may be a function of not only the system’s energy E, but also of its volume: S=S(E,V). Let us consider this relation resolved for energy: E=E(S,V), and write the general mathematical expression for the full differential of E as a function of these two independent arguments:
dE=(∂E∂S)VdS+(∂E∂V)SdV.
This formula, based on the stationary relation E=E(S,V), is evidently valid not only in equilibrium but also for all very slow, reversible21 processes. Now, using Eqs. (1.2.6) and (???), we may rewrite Equation (???) as
Energy: differential dE=TdS−PdV.
According to Equation (1.1.1), the second term on the right-hand side of this equation is just the work of the external force, so that due to the conservation of energy,22 the first term has to be equal to the heat dQ transferred from the environment to the system (see Figure 1.3.1):
1st law of thermodynamics:
dE=dQ+dW,
dQ=TdS.
The last relation, divided by T and then integrated along an arbitrary (but reversible!) process,
S=∫dQT+const,
is sometimes used as an alternative definition of entropy S – provided that temperature is defined not by Equation (1.2.6), but in some independent way. It is useful to recognize that entropy (like energy) may be defined to an arbitrary constant, which does not affect any other thermodynamic observables. The common convention is to take
S→0, at T→0.
This condition is sometimes called the “3rd law of thermodynamics”, but it is important to realize that this is just a convention rather than a real law.23 Indeed, the convention corresponds well to the notion of the full order at T=0 in some systems (e.g., separate atoms or perfect crystals), but creates ambiguity for other systems, e.g., amorphous solids (like the usual glasses) that may remain highly disordered for “astronomic” times, even at T→0.
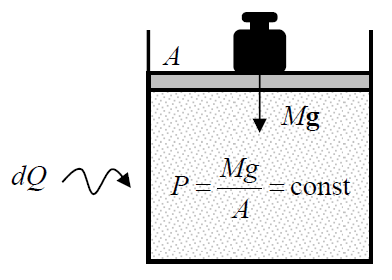
Now let us discuss the notion of heat capacity that, by definition, is the ratio dQ/dT, where dQ is the amount of heat that should be given to a system to raise its temperature by a small amount dT.24 (This notion is important because the heat capacity may be most readily measured experimentally.) The heat capacity depends, naturally, on whether the heat dQ goes only into an increase of the internal energy dE of the system (as it does if its volume V is constant), or also into the mechanical work (–dW) performed by the system at its expansion – as it happens, for example, if the pressure P, rather than the volume V, is fixed (the so-called isobaric process – see Figure 1.3.2).
Hence we should discuss at least two different quantities,25 the heat capacity at fixed volume,
Cv≡(∂Q∂T)V
and the heat capacity at fixed pressure
Cp≡(∂Q∂T)P,
and expect that for all “normal” (mechanically stable) systems, CP≥CV. The difference between CP and CV is rather minor for most liquids and solids, but may be very substantial for gases – see Sec. 4.