7.6: The Quantum Harmonic Oscillator
- Page ID
- 4531

By the end of this section, you will be able to:
- Describe the model of the quantum harmonic oscillator
- Identify differences between the classical and quantum models of the harmonic oscillator
- Explain physical situations where the classical and the quantum models coincide
Oscillations are found throughout nature, in such things as electromagnetic waves, vibrating molecules, and the gentle back-and-forth sway of a tree branch. In previous chapters, we used Newtonian mechanics to study macroscopic oscillations, such as a block on a spring and a simple pendulum. In this chapter, we begin to study oscillating systems using quantum mechanics. We begin with a review of the classic harmonic oscillator.
The Classic Harmonic Oscillator
A simple harmonic oscillator is a particle or system that undergoes harmonic motion about an equilibrium position, such as an object with mass vibrating on a spring. In this section, we consider oscillations in one-dimension only. Suppose a mass moves back-and-forth along the \(x\)-direction about the equilibrium position, \(x = 0\). In classical mechanics, the particle moves in response to a linear restoring force given by \(F_x = -kx\), where \(x\) is the displacement of the particle from its equilibrium position. The motion takes place between two turning points, \(x \pm A\), where A denotes the amplitude of the motion. The position of the object varies periodically in time with angular frequency \(\omega = \sqrt{k/m}\), which depends on the mass m of the oscillator and on the force constant \(k\) of the net force, and can be written as
\[x(t) = A \, \cos (\omega t + \phi). \label{7.52} \]
The total energy \(E\) of an oscillator is the sum of its kinetic energy \(K = mu^2/2\) and the elastic potential energy of the force \(U(x) = kx^2/2\),
\[E = \dfrac{1}{2} mu^2 + \dfrac{1}{2}kx^2. \label{7.53} \]
At turning points \(x = \pm A\), the speed of the oscillator is zero; therefore, at these points, the energy of oscillation is solely in the form of potential energy \(E = kA^2/2\). The plot of the potential energy \(U(x)\) of the oscillator versus its position \(x\) is a parabola (Figure \(\PageIndex{1}\)). The potential-energy function is a quadratic function of \(x\), measured with respect to the equilibrium position. On the same graph, we also plot the total energy \(E\) of the oscillator, as a horizontal line that intercepts the parabola at \(x = \pm A\). Then the kinetic energy \(K\) is represented as the vertical distance between the line of total energy and the potential energy parabola.
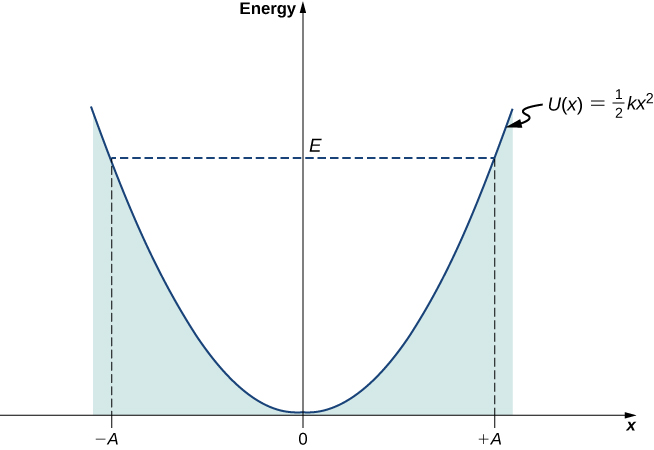
In this plot, the motion of a classical oscillator is confined to the region where its kinetic energy is nonnegative, which is what the energy relation Equation \ref{7.53} says. Physically, it means that a classical oscillator can never be found beyond its turning points, and its energy depends only on how far the turning points are from its equilibrium position. The energy of a classical oscillator changes in a continuous way. The lowest energy that a classical oscillator may have is zero, which corresponds to a situation where an object is at rest at its equilibrium position. The zero-energy state of a classical oscillator simply means no oscillations and no motion at all (a classical particle sitting at the bottom of the potential well in Figure \(\PageIndex{1}\)). When an object oscillates, no matter how big or small its energy may be, it spends the longest time near the turning points, because this is where it slows down and reverses its direction of motion. Therefore, the probability of finding a classical oscillator between the turning points is highest near the turning points and lowest at the equilibrium position. (Note that this is not a statement of preference of the object to go to lower energy. It is a statement about how quickly the object moves through various regions.)
The Quantum Harmonic Oscillator
One problem with this classical formulation is that it is not general. We cannot use it, for example, to describe vibrations of diatomic molecules, where quantum effects are important. A first step toward a quantum formulation is to use the classical expression \(k = m\omega^2\) to limit mention of a “spring” constant between the atoms. In this way the potential energy function can be written in a more general form,
\[U(x) = \dfrac{1}{2}m \omega^2 x^2. \label{7.54} \]
Combining this expression with the time-independent Schrӧdinger equation gives
\[-\dfrac{\hbar}{2m} \dfrac{d^2 \psi(x)}{dx^2} + \dfrac{1}{2}m\omega^2 x^2 \psi(x) = E\psi (x). \label{7.55} \]
To solve Equation \ref{7.55}, that is, to find the allowed energies \(E\) and their corresponding wavefunctions \(\psi (x) \) - we require the wavefunctions to be symmetric about \(x = 0\) (the bottom of the potential well) and to be normalizable. These conditions ensure that the probability density \(|\psi (x)|^2\) must be finite when integrated over the entire range of x from \(-\infty\) to \(+\infty\). How to solve Equation \ref{7.55} is the subject of a more advanced course in quantum mechanics; here, we simply cite the results. The allowed energies are
\[ \begin{align} E_n &= \left(n + \dfrac{1}{2}\right) \hbar \omega \\[5pt] &= \dfrac{2n + 1}{2} \hbar \omega \label{7.56} \end{align} \]
with \(n = 0,1,2,3,...\)
The wavefunctions that correspond to these energies (the stationary states or states of definite energy) are
\[\psi_n (x) = N_n e^{-\beta^2 x^2/2} H_n (\beta x), \, n = 0,1,2,3, ... \label{7.57} \]
where \(\beta = \sqrt{m\omega/\hbar}\), \(N_n\) is the normalization constant, and \(H_n(y)\) is a polynomial of degree \(n\) called a Hermite polynomial. The first four Hermite polynomials are
- \(H_0 (y) = 1\)
- \(H_1 (y) = 2y\)
- \(H_2 (y) = 4y^2 - 2\)
- \(H_3 (y) = 8y^3 - 12 y.\)
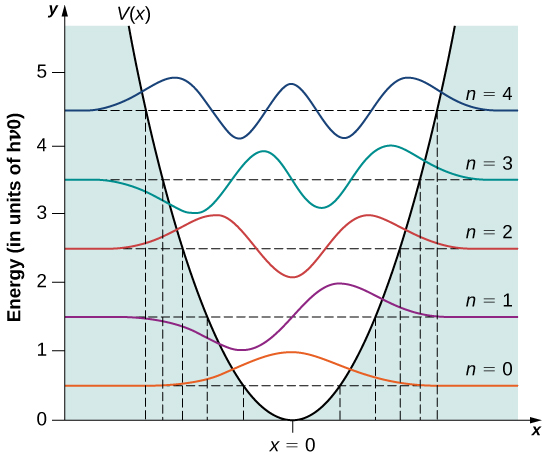
A few sample wavefunctions are given in Figure \(\PageIndex{2}\). As the value of the principal number increases, the solutions alternate between even functions and odd functions about \(x = 0\).
Find the amplitude \(A\) of oscillations for a classical oscillator with energy equal to the energy of a quantum oscillator in the quantum state \(n\).
Strategy
To determine the amplitude \(A\), we set the classical energy \(E = kx^2/2 = m\omega^2 \, A^2/2\) equal to \(E_n\) given by Equation \ref{7.56}.
Solution
We obtain
\[\begin{align} E_n &= m\omega^2 A_n^2/2 \nonumber \\[5pt] A_n &= \sqrt{\dfrac{2}{m\omega^2}E_n} \nonumber \\[5pt] &= \sqrt{\dfrac{2}{m\omega^2} \dfrac{2n + 1}{2}\hbar \omega} \nonumber \\[5pt] &= \sqrt{(2n + 1) \dfrac{\hbar}{m\omega}}. \nonumber \end{align} \nonumber \]
Significance
As the quantum number n increases, the energy of the oscillator and therefore the amplitude of oscillation increases (for a fixed natural angular frequency. For large n, the amplitude is approximately proportional to the square root of the quantum number.
Several interesting features appear in this solution. Unlike a classical oscillator, the measured energies of a quantum oscillator can have only energy values given by Equation \ref{7.56}. Moreover, unlike the case for a quantum particle in a box, the allowable energy levels are evenly spaced,
\[\begin{align} \Delta E &= E_{n+1} - E_n \\[5pt] &= \dfrac{2(n + 1) + 1}{2} \hbar \omega - \dfrac{2n + 1}{2} \hbar \omega \\[5pt] &= \hbar \omega = hf. \label{7.58} \end{align} \]
When a particle bound to such a system makes a transition from a higher-energy state to a lower-energy state, the smallest-energy quantum carried by the emitted photon is necessarily \(hf\). Similarly, when the particle makes a transition from a lower-energy state to a higher-energy state, the smallest-energy quantum that can be absorbed by the particle is \(hf\). A quantum oscillator can absorb or emit energy only in multiples of this smallest-energy quantum. This is consistent with Planck’s hypothesis for the energy exchanges between radiation and the cavity walls in the blackbody radiation problem.
The \(\ce{HCl}\) diatomic molecule consists of one chlorine atom and one hydrogen atom. Because the chlorine atom is 35 times more massive than the hydrogen atom, the vibrations of the \(\ce{HCl}\) molecule can be quite well approximated by assuming that the Cl atom is motionless and the H atom performs harmonic oscillations due to an elastic molecular force modeled by Hooke’s law. The infrared vibrational spectrum measured for hydrogen chloride has the lowest-frequency line centered at \(f = 8.88 \times 10^{13} Hz\). What is the spacing between the vibrational energies of this molecule? What is the force constant k of the atomic bond in the HCl molecule?
Strategy
The lowest-frequency line corresponds to the emission of lowest-frequency photons. These photons are emitted when the molecule makes a transition between two adjacent vibrational energy levels. Assuming that energy levels are equally spaced, we use Equation \ref{7.58} to estimate the spacing. The molecule is well approximated by treating the Cl atom as being infinitely heavy and the H atom as the mass \(m\) that performs the oscillations. Treating this molecular system as a classical oscillator, the force constant is found from the classical relation \(k = m\omega^2\).
Solution
The energy spacing is
\[ \begin{align} \Delta E &= hf \nonumber \\[5pt] &= (4.14 \times 10^{-15} eV \cdot s)(8.88 \times 10^{13} Hz) \nonumber\\[5pt] &= 0.368 \, eV. \nonumber \end{align} \nonumber \]
The force constant is
\[ \begin{align} k &= m \omega^2 \nonumber \\[5pt] &= m (2\pi f)^2 \nonumber \\[5pt] &= (1.67 \times 10^{ −27} kg)(2\pi \times 8.88 \times 10 ^{13}Hz)^2 \nonumber \\[5pt] &= 520 \, N/m. \nonumber \end{align} \nonumber \]
Significance
The force between atoms in an HCl molecule is surprisingly strong. The typical energy released in energy transitions between vibrational levels is in the infrared range. As we will see later, transitions in between vibrational energy levels of a diatomic molecule often accompany transitions between rotational energy levels.
The vibrational frequency of the hydrogen iodide HI diatomic molecule is \(6.69×10^{ 13}\,Hz\).
- What is the force constant of the molecular bond between the hydrogen and the iodine atoms?
- What is the energy of the emitted photon when this molecule makes a transition between adjacent vibrational energy levels?
- Answer a
-
295 N/m
- Answer b
-
0.277 eV
The quantum oscillator differs from the classic oscillator in three ways:
- First, the ground state of a quantum oscillator is \(E_0 = \hbar \omega /2\), not zero. In the classical view, the lowest energy is zero. The nonexistence of a zero-energy state is common for all quantum-mechanical systems because of omnipresent fluctuations that are a consequence of the Heisenberg uncertainty principle. If a quantum particle sat motionless at the bottom of the potential well, its momentum as well as its position would have to be simultaneously exact, which would violate the Heisenberg uncertainty principle. Therefore, the lowest-energy state must be characterized by uncertainties in momentum and in position, so the ground state of a quantum particle must lie above the bottom of the potential well.
- Second, a particle in a quantum harmonic oscillator potential can be found with nonzero probability outside the interval \(-A \leq x \leq +A\). In a classic formulation of the problem, the particle would not have any energy to be in this region. The probability of finding a ground-state quantum particle in the classically forbidden region is about 16%.
- Third, the probability density distributions \(|\psi_n(x)|^2\) for a quantum oscillator in the ground low-energy state, \(\psi_0(x)\), is largest at the middle of the well \((x = 0)\). For the particle to be found with greatest probability at the center of the well, we expect that the particle spends the most time there as it oscillates. This is opposite to the behavior of a classical oscillator, in which the particle spends most of its time moving with relative small speeds near the turning points.
Find the expectation value of the position for a particle in the ground state of a harmonic oscillator using symmetry.
- Answer b
-
\[\langle x \rangle = 0 \nonumber \]
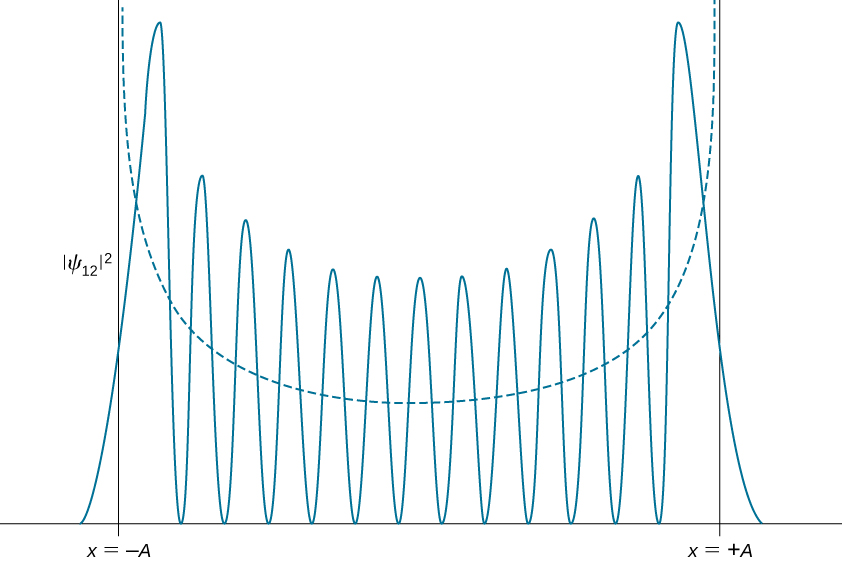
Quantum probability density distributions change in character for excited states, becoming more like the classical distribution when the quantum number gets higher. We observe this change already for the first excited state of a quantum oscillator because the distribution \(|\psi_1(x)|^ 2\) peaks up around the turning points and vanishes at the equilibrium position, as seen in Figure \(\PageIndex{2}\). In accordance with Bohr’s correspondence principle, in the limit of high quantum numbers, the quantum description of a harmonic oscillator converges to the classical description, which is illustrated in Figure \(\PageIndex{3}\). The classical probability density distribution corresponding to the quantum energy of the \(n = 12\) state is a reasonably good approximation of the quantum probability distribution for a quantum oscillator in this excited state. This agreement becomes increasingly better for highly excited states.