
6.3: Entropy
- Page ID
- 18481

Extensive and Intensive State Variables
All state variables can be classified into one of two categories, which we call extensive and intensive. Extensive state variables are those that are additive when two systems are combined into one, while intensive state variables are not additive. For example, suppose we have two identical boxes of the same gas in the same thermodynamic state that are separated by a barrier. Removing the barrier and treating the new bigger box as a single system, we find that we have doubled the volume, particle number, and internal energy of the system. These are therefore extensive state variables. But if we measure the pressure or temperature of the new state, we find that these quantities are unchanged, making them intensive variables. All state variables come in one variety or the other.
The way to tell mathematically whether a state variable is extensive or intensive is to look at what happens to it as a function of other state variables when they are scaled according to whether they are extensive or not. So for example, suppose we know that the number of moles \(n\) is extensive and the total energy \(U\) in an ideal gas system are extensive, but we are not sure about temperature \(T\). Writing temperature in terms of \(n\) and \(U\) gives:
\[T\left(n,U\right)=\dfrac{U}{nC_V}\]
Scaling the system by a factor of \(k\) (i.e. putting together \(k\) identical systems) changes \(n\) and \(U\) by that factor but has no effect on \(T\):
\[T\left(k\cdot n,k\cdot U\right)=\dfrac{k\cdot U}{k\cdot nC_V}=\dfrac{U}{nC_V}=T\left(n,U\right)\]
Example \(\PageIndex{1}\)
Given that \(n\) and \(V\) are extensive, and \(T\) is intensive, use the ideal gas law to show that \(P\) is intensive.
- Solution
-
Following the same process as above where we scale the extensive variables (\(n\rightarrow k\cdot n\), \(V\rightarrow k\cdot V\)), and do nothing to the intensive variables (\(T\rightarrow T\)) we have:
\[P\left(k\cdot n,k\cdot V,T\right)=\dfrac{k\cdot nRT}{k\cdot V} = \dfrac{nRT}{V} = P\left(n,V,T\right)\nonumber\]
Since \(P\) doesn't change upon scaling the system, it is an intensive state variable.
Whether a variable is intensive or extensive also affects the final result when we combine two systems that are not identical. If we again consider two systems separated by a barrier which this time have different state variables (but are the same type of gas), then we get the following differences in the results for each type of variable:
Figure 6.3.1 – State Variables in Combined Systems
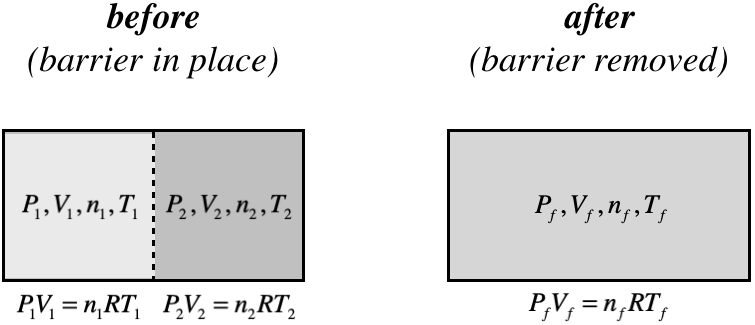
The extensive variables will simply add. This is obvious for particle number and volume, but the sum of the internal energies of the two systems must equal the internal energy of the combined system as well (energy isn't created or destroyed by the combination):
\[V_f=V_1+V_2\;,\;\;\;\;n_f=n_1+n_2\;,\;\;\;\;U_f=U_1+U_2\]
We can use these to determine what happens to the intensive variables. First use the internal energy relation to determine the fate of the temperature:
\[n_fC_VT_f = n_1C_VT_1 + n_2C_VT_2 \;\;\;\Rightarrow\;\;\; T_f = \dfrac{n_1T_1 + n_2T_2}{n_1 + n_2}\]
And combining this with the ideal gas law gives a similar result for pressure:
\[P_fV_f=n_fRT_f \;\;\;\Rightarrow\;\;\; P_f\left(V_1+V_2\right) = \left(n_1 + n_2\right)R\left[\dfrac{n_1T_1 + n_2T_2}{n_1 + n_2}\right]\;\;\;\Rightarrow\;\;\;P_f=\dfrac{V_1P_1 + V_2P_2}{V_1 + V_2}\]
So you see that the temperature and pressure of the combined system are "weighted averages" of the temperatures and pressures of the individual systems.
A New State Variable
We know that work and heat are similar in that they are the two quantities in thermodynamics that are not state variables. We know specifically how work arises from a single infinitesimal step in a quasi-static process: \(dW = PdV\), but as yet we have no equivalent relation for heat. If we assume, because of its similarity with work, that heat does have such a relation with two state functions, then it should look something like: \(dQ = XdY\). Comparing this with the case of work, and keeping in mind what properties “drive” work and heat, which of the following are likely candidates for the state functions \(X\) and \(Y\)?
Well, we know that a pressure difference on the two sides of a piston will result in work being done, and while work is "driven" by a pressure difference, heat is driven by a temperature difference. We therefore infer that the state variable \(X\) is temperature, but \(Y\) is a mystery. One thing we can perhaps conclude that since work is defined from a combination of an intensive function (\(P\)) and the change of an extensive function (\(dV\)), then \(dY\) should be extensive (note that the driving variable temperature is intensive, as pressure is in the case of work).
None of the state variables we have seen so far fit the bill, so we'll just postulate the existence of another state variable we call entropy, and give it the symbol \(S\). We therefore have, in analogy with work the relation for a small quantity of heat:
\[dQ = TdS\]
The units for entropy are the same as those for heat capacity (\(\frac{J}{K}\)), but heat capacity and entropy are not the same. As with the case of work, we can add up all the small contributions to heat transferred during a quasi-static process:
This is good place to point out that these relationships for work and heat can also be turned around to result in a change of a state function for a quasi-static process:
\[\begin{array}{l} dV = \dfrac{dW}{P} && \Rightarrow && \Delta V = V_B-V_A && = && \int\limits_A^B\dfrac{dW}{P} \\ dS = \dfrac{dQ}{T} && \Rightarrow && \Delta S = S_B-S_A && = && \int\limits_A^B\dfrac{dQ}{T} \end{array}\]
Of course, the first of these relations is virtually never used, because we can generally just look at what the piston does to determine the volume change – we don’t have to calculate it. The second relation is another matter – the change in entropy is not immediately apparent, and as we will see, knowing the change of entropy can be quite important to understanding a process.
Question: A gas is sealed in an insulated cylinder with a piston. The piston is then compressed slowly (a quasi-static process), and the temperature of the gas rises. In what direction does the entropy function change for that gas?
Answer: The piston is insulated, which means that the gas does not exchange heat with the outside environment. This is a quasi-static process, so \(dQ = 0\) throughout the process means that \(dS = 0\) throughout the process, which means that \(\Delta S = 0\).
Here we can see why the \(\Delta S\) equation is so much more commonly-used than the \(\Delta V\) equation – we know that an adiabatic process occurs when we insulate the container, and that this prevents heat from being transferred, so the entropy change is zero. The equivalent process for the case of work is the isochoric process, which we know immediately involves zero change in volume – we don’t need to reason that we have rigged the apparatus so that no work is exchanged and therefore conclude that there must not have been a volume change.
In the case of a quasi-static process where zero work is done, we were able to characterize it in terms of a changing state variable – an isochoric process. Until now, we had no such characterization for a quasi-static process where no heat is exchanged. But so long as we are talking about a quasi-static process, we can rename the adiabatic process in terms of the unchanging state variable: an isentropic process.
Ideal Gases
When we first started discussing state functions, we said that there are four independent state variables needed to define a state, but when we know more about how the particles interact (such as an ideal gas, where they don't interact), this number drops to three. We said that this allows us to express one state variable in terms of three others, as we did in Equation 5.5.2. Well, now we have a new state variable, and it is useful to express it as a function of three others. Derivation of this expression is a bit involved, but here it is:
\[S\left(N,U,V\right) = Nk_B\ln\left[\left(\dfrac{V}{N}\right)\left(\dfrac{U}{N}\right)^{\dfrac{1}{\gamma-1}}\right]\]
One thing to note here is that while the ideal gas law state equation is the same for all ideal gases, the state equation for entropy depends upon the type (monatomic, diatomic, etc) of ideal gas, as evidenced by the presence of the constant \(\gamma\).
This beastly-looking expression actually carries with it quite a lot of power, though it can take a bit of mathematics to extract it. We can get more by rewriting the entropy in terms of three other variables. For example, we can replace internal energy with pressure by noting that:
\[U = nC_VT = \dfrac{C_V}{R}nRT = \dfrac{C_V}{R}PV = \dfrac{1}{\gamma - 1}PV\]
Using the properties of the logarithm and doing the algebra to simplify the entropy function gives:
\[S\left(N,P,V\right)=\dfrac{Nk_B}{\gamma - 1} \ln\left[PV^{\gamma}\right] + f\left(N\right)\;,\]
where \(f\left(N\right)\) is a function of \(N\) that does not need to be written out here. Holding \(N\) constant as we always do in our limited treatment of thermodynamics, we can write this equation as:
\[S\left(N,P,V\right)=\left(constant\right) \ln\left[PV^{\gamma}\right] + \left(constant\right)\]
If we now ask, “On what curve on a \(PV\) graph will the entropy of the state remain constant (\(\Delta S = 0\))?”, the answer is obvious:
\[\Delta S = 0 \text{ when } PV^{\gamma} = constant\]
This is the equation for an adiabat, which is a curve for which there is no heat transfer when the process is quasi-static. This makes sense, given the relationship between entropy and heat exchange.
Entropy Changes in Special Processes
Back when we studied the four “special” processes, we derived all the changes in state functions for each process. We can do the same for entropy using the function above, but it is simpler to do with the integral. We have already stated (and shown) that for a quasi-static adiabatic process the entropy change is zero. Let’s see how we get the change for the other processes. In every case, we are using Equation 6.3.7, and plugging in what we know about heat for each case from Section 5.8 (note that the final equality in each result uses the ideal gas law and the constant variable in that case):
Free Expansion
We ended Section 5.8 with an important discussion about how we can deal with non-quasi-static processes even though all of our models and results regarding processes are centered around solving quasi-static processes. The subtle and important point is that if the endpoints of such a process are equilibrium states, then the values of the state variables at those points are well-defined, and we can sometimes use what we know about the process to relate those points to each other, and then use what we know about special processes to solve for something useful. What follows is undoubtedly the best example of this method.
Consider an insulated container of a monatomic ideal gas that has all of the gas confined at equilibrium in one half of the container, thanks to a membrane (barrier) between the two halves, with the other half completely evacuated. Suddenly, the membrane ruptures, and gas quickly fills the container and eventually returns to equilibrium. We call this process free expansion. It helps to put this process into our usual context of a gas confined with a piston, so here is an appropriate picture of what is going on:
Figure 6.3.2 – Free Expansion as a Sudden Shove of a Piston
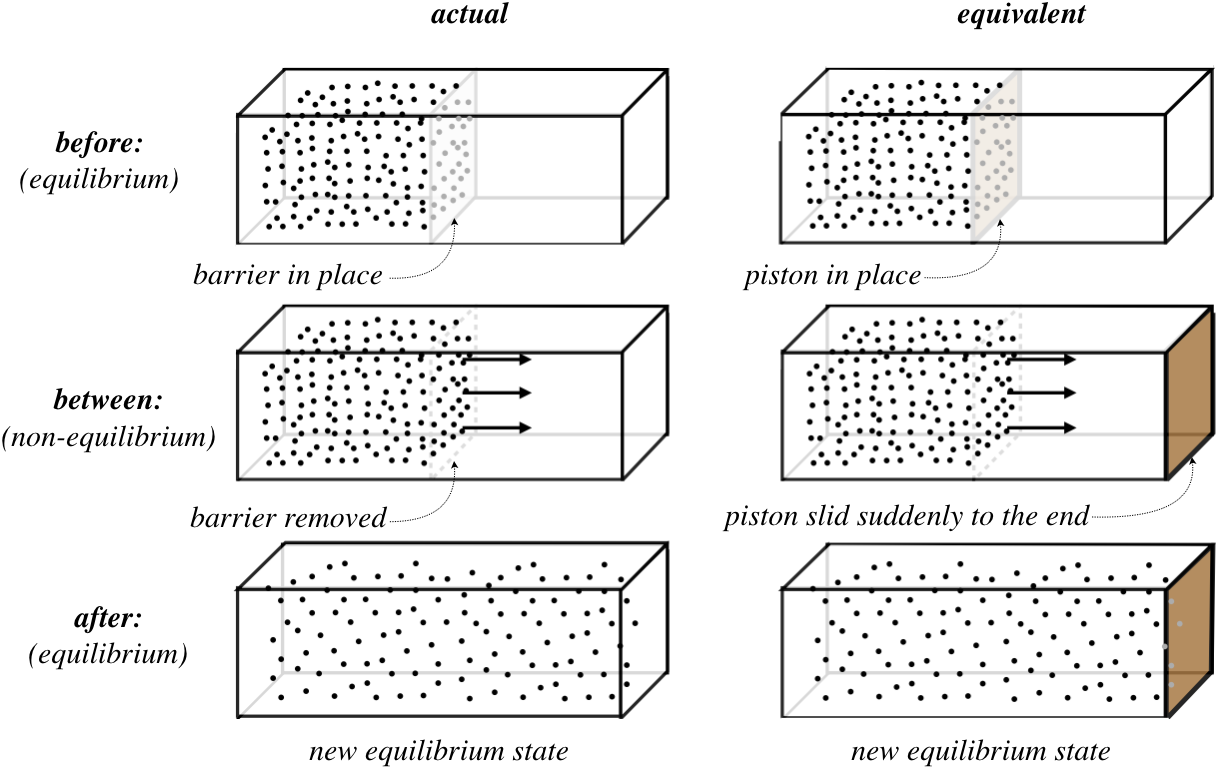
Clearly this is not a quasi-static process. But that doesn't mean we can't say anything about how the beginning and ending states are related. We are defining our system as the whole container, and it is insulated, so we know for sure that no heat enters the system while it evolves. Also, the piston is shoved so quickly that the gas particles do not rebound against it as it moves. Consequently, the gas exerts no force on the piston, and therefore doesn't do any work. [That is not to say that no work is done at all, of course – we do work on the piston from outside the system. But the point is that the work done on the piston is not reflected in the state of the gas.]
With no work done on or by the gas, and no heat added to or taken away from the gas, the first law ensures that the internal energy of the gas is unchanged. This actually makes sense, since the internal energy is the sum of the kinetic energies of all the particles, and why would suddenly removing the barrier cause any of the particles to change the kinetic energies they had just before the barrier moved? But when one considers that an unchanging internal energy means that the temperature is also unchanged, then it starts to seem a bit weird – we are used to quick expansions cooling the gas. But here we have to be careful. A gas cools when it expands adiabatically, which assumes that work is done on the piston. We only treated "quick" expansion as adiabatic because it was a practical means of having an expansion without significant heat loss. But free expansion is not the same as adiabatic expansion – there is no work done on a piston at all, and therefore no way for the internal energy to exit the system.
So with no work done, no heat transferred, and no change in temperature, what has changed? Well, clearly the volume has doubled. With the temperature and number of particles unchanged, the ideal gas law tells us that the pressure is cut in half. That accounts for all of the state variables except for one... What about the entropy? It is tempting to say that because there is no heat exchanged that \(Q=\int TdS\) tells us that the entropy change is also equal to zero, but this is not correct! The reason is that this relation, like \(W=\int PdV\), only links two equilibrium states when a quasi-static process is followed, and this is not such a process.
We therefore turn to our trick – we use what we know about the actual process and the equilibrium endpoints to invent any quasi-static process whatsoever between the two endpoints to calculate the entropy change. To see how we might do this, let's start by plotting the two points on a \(PV\) diagram:
Figure 6.3.3 – PV diagram of Free Expansion
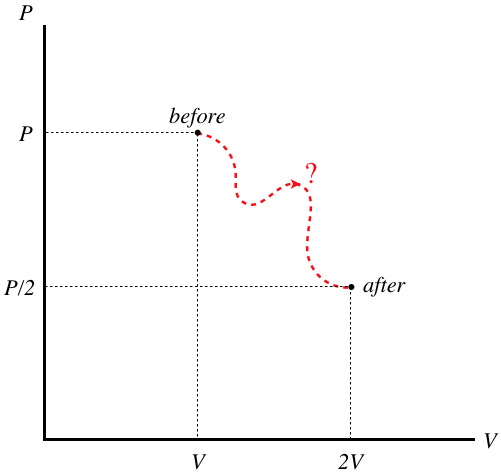
There is no "correct" path here – the system does not pass through any equilibrium states during its journey. But as we are looking for a change in a state function, only the endpoints matter, and if a particular path is useful, we can go ahead and use it. So let's just run through our options one-by-one. Both the pressure and volume are changing, so neither an isochoric nor an isobaric process will connect these dots. Let's try an adiabat. in this case, the pressure and volume are related in a specific way, so we can check to see if the endpoints lie along the same adiabat:
\[P_1V_1^{\gamma} = P_2V_2^{\gamma} \;\;\;\Rightarrow\;\;\; PV^{\gamma} = \left(\dfrac{P}{2}\right)\left(2V\right)^{\gamma} \;\;\;\Rightarrow\;\;\; PV^{\gamma} \ne 2^{\gamma-1}PV^{\gamma}\]
The two points do not lie along an adiabat. Wait a minute – we already said that the temperature doesn't change, so we know these points lie along an isotherm. We already have this solution in terms of the volume (or pressure) change from Equation 6.3.14, so we get the answer immediately:
\[\Delta S = nR\ln\left[\dfrac{V_B}{V_A}\right] = nR\ln\left[\dfrac{2V}{V}\right] = nR\ln 2\]
Example \(\PageIndex{2}\)
For the free-expansion case above, show that you can get the same entropy change, even if you choose the less-convenient path between the endpoints that is first isochoric to the proper pressure, and then isobaric to the proper volume.
- Solution
-
The first process is isochoric from \(P\) to \(\frac{P}{2}\), and we have the entropy change for this process in Equation 6.3.14:
\[\Delta S_1 = nC_V\ln\left[\dfrac{P_B}{P_A}\right] = nC_V\ln\left[\dfrac{\frac{P}{2}}{P}\right] = nC_V\ln\left[\frac{1}{2}\right]=-nC_V\ln 2\nonumber\]
The second process is isobaric from \(V\) to \(2V\), and once again we have already computed this result in Equation 6.3.14:
\[\Delta S_2 = nC_P\ln\left[\dfrac{V_A}{V_B}\right] = nC_P\ln\left[\dfrac{2V}{V}\right] = nC_P\ln 2=nC_P\ln 2\nonumber\]
The total change in entropy is the sum of these changes:
\[\Delta S = -nC_V\ln 2 + nC_P\ln 2 = n\left(C_P-C_V\right)\ln 2 = nR\ln 2\nonumber\]
Example \(\PageIndex{3}\)
For the free-expansion case above, show that you can get the same entropy change using Equation 6.3.11.
- Solution
-
Noting that the particle number doesn't change and computing the entropy change directly using properties of the logarithm:
\[\Delta S = S_2 - S_1=\dfrac{Nk_B}{\gamma - 1} \left\{\ln\left[P_2V_2^{\gamma}\right] - \ln\left[P_1V_1^{\gamma}\right]\right\} = \dfrac{nR}{\gamma - 1} \left\{\ln\left[\left(\frac{1}{2}P\right)\left(2V\right)^{\gamma}\right] - \ln\left[PV^{\gamma}\right]\right\} = \dfrac{nR}{\gamma - 1}\ln\left[2^{\gamma-1}\right]=nR\ln 2\nonumber\]