
7.3: Molecular Dynamics for Biomembranes


- Last updated
- Mar 28, 2021
- Save as PDF
( \newcommand{\kernel}{\mathrm{null}\,}\)
Molecular dynamics (MD) is a simulation method used in many fields to help understand the movement of atoms and molecules. This method is based on Newton's equation of motion and generally does not include quantum mechanics. Each atom or molecule in the simulation is defined by a force field and mass, and is allowed to interact for a given time period. These time periods are kept short due to error propagation after many approximations. Many of the problems that use molecular dynamics often have a large number of atoms and are analytically intractable. Incorporation of boundary conditions allows researchers to reduce the size of a simulation without sacrificing pertinent information. One can run the desired simulation on the smallest possible unit, and surround it with replicas extending infinitely in all directions. This results in a “bigger” simulation that still requires minimal computation.
Biomembranes play a tremendously important role in organisms, separating cells from their environment and compartmentalizing organelles with unique and individual functions. Membranes contribute to the structure of cells and organelles via specific lipid composition and control what molecules cross the membrane through networks of protein channels interspersed among the lipid groups. Given the many important roles of biomembranes, understanding their composition, structure, and interactions with intracellular material and the environment is important for drug discovery and delivery. Because of the unique electrostatic and chemical properties of biomembranes, it is difficult and laborious to visualize them experimentally in a way that reflects their dynamic nature. For this reason, molecular dynamic simulations have become a ubiquitous method of visualizing membrane behavior and interactions with molecules of interest.
Physical Basis: Newton's Second Law
Molecular dynamics (MD) utilize equations from classical mechanics to describe the way in which atoms and small molecules interact. Quantum equations and relativity are ignored, but these exclusions minimally impact the implications of the tests being run. Approximations based on classical mechanics are usually sufficient to make accurate conclusions. MD is centered around Newton’s second law of motion. Rather than the traditional notation, this equation is expressed as a gradient of potential energy1:
→F1=−∇iV(¯r3})
This equation expresses the gradient with respect to the position of the particle, i. F represents the conservative forces between atoms in the system, −∇iV is the gradient of the potential energy, and is the position of atoms. This equation can be adjusted to express the acceleration using F=ma, where the acceleration is the second time derivative of position:
m∂2t→ri=−∇iV(→{rj})
The right hand of this calculation is effectively mass times acceleration for Newton’s second law. In biomembrane systems, statistical and thermodynamical properties will reduce Hamilton's equations to the same equation above1.
Using the Verlet Integrator to solve Newton's Second Law
While executing MD simulations, an integrator is applied to this partial differential equation to calculate positions and velocities of molecules based on initial conditions. There are many types of integrators commonly used by researchers. Most are based on the Taylor expansion of positions as a function of time:
→r(t)=∞∑i=0∂it→rt=t0(t−t0)ii!
A common integrator is the Verlet integrator, which expands the above equation to the third order. The first derivative describes the velocity of a particle, the second is acceleration, and the third is jerk. The equation is performed positively and negatively with respect to time in order to maintain the symmetry necessary for accurate statistical mechanical description2:
Forward Expansion
→r(t)=→r(t0)+→v(t0)Δt+→a(t0)Δt22+∂3t→r(t0)Δt36+O(Δt4)
Backward Expansions
→r(t)=→r(t0)−→v(t0)Δt+→a(t0)Δt22−∂3t→r(t0)Δt36+O(Δt4)
In these equations, acceleration is represented by a. O is a constant used to abbreviate the fourth order Taylor expansion of the position equation. These equations are then combined to give the position algorithm expanded to the fourth order. This equation allows us to predict the position of all atoms in a system given we know the preceding and subsequent timesteps:
→r(t0+Δt)=2→r(t0)−→r(t0−Δt)+→a(t0)Δt2+O(Δt4)
The Isobaric-Isothermal Ensemble
To simulate common biomembrane environments, an isobaric-isothermal ensemble is used20. A statistical ensemble is a large set of replicated microstates that have the same macrostate. This is analogous to a scientist using the same conditions to test systems which may not be microscopically identical.
The isobaric-isothermal ensemble is also called the NPT ensemble, in which the number of particles N is conserved, and a constant pressure P and constant temperature T are applied. This mimics common biomembrane environments: liquids at constant temperature with the ability for lipids and proteins to expand and contract.
The Berendsen barostat is a common tool to maintain the effective pressure of the system. This is done by re-scaling box size and particle position to restore a target pressure (ptarget). This is done using the following equation1:
V new Vold=Δtτ(1+pptarget)
In this equation, tau indicates the frequency with which particle positions are re-adjusted. Vnew and Vold indicate the box volumes at two adjacent timesteps.
The Nose-Hoover thermostat controls the kinetic degrees of freedom in order to keep the temperature constant22. Using a many-body Hamiltonian and a time-derivative scaling factor, the isothermal equations of motion can be expressed as multiple equations of motion where s is the scaling factor, Q is the position, P is the momentum, and each is a first order derivative:
˙Qi=Pi/mis,˙Pi=sFi
˙s=sαPs,˙Ps=∑i[P2imis2−DkBT]
This method is deterministic, time-reversible, and easy to implement. The problem is that not every system will be ergodic (the probability average will not be the long-term average).
Modeling a Membrane
MD allows us to monitor the movement and interactions of individual atoms within a molecule. This process is time consuming and computationally intensive, which limits the time-scale and area over which such calculations can be executed. In a biomembrane model, modeling the chemically interesting water molecules alone would take far longer than is practical, and only a few lipid groups would be able to be modeled. Viewing a whole unit of interest this way would be limited to a nanosecond simulation time. Many all-atom simulations of bilayers only model 128 lipids, or 64 on each layer1. This style of simulations has a number of useful applications, including looking at local structural characteristics and dynamics of membranes as well as how specific components such as sterols affect local membrane structure. However, events such as bilayer assembly and phase transitions and separations occur on time-scales unreasonable for atomistic models.
Using too big of a timestep might miss important transitions that affect the properties of the system, but too short of a timestep is computationally expensive. Angle vibrations are on the order of 10 femtoseconds, and it is advisable to make the timestep an order of magnitude shorter than the smallest observed events1. Generally 1 femtosecond to a few picoseconds is an acceptable range.
Parameters used for Biomembranes
The math utilized for each parameter varies by program, but the functions will be summarized here briefly.
Bond angles and bond lengths: Bond angles and lengths are typically constrained to biologically relevant values. Each residue has a range of angles it is expected to adopt, and these expectations are implemented into a model’s parameters. Whether or not bonds are allowed to stretch or oscillate between different angles is a controllable parameter.
Torsional degrees of freedom: Torsional degrees of freedom are the different bond angles at which a molecule can be stable. For example, in CO2 the molecule is linear, so there are no different bond angle configurations it may take. In n-butane, which takes the form of CH3-CH2-CH2-CH3, where each of those bonds can take 3 energetic positions. This parameter is regularly constrained.
Chain order parameter: Chain order is a measure of the angle of tail C-H bond and the bilayer normal. This can be defined for a single lipid1. The order parameter can be calculated using the following equation (S is the order parameter, and Theta CD is the angle between the C-H bond and the bilayer normal):
ScD=0.5⟨3cos2Θcp−1⟩
The order parameter is calculated for all saturated carbons. Unsaturated carbons no longer have a tetrahedral orientation and the order parameter does not apply.
Effects of parameters
The basic premise of MD is building an equation or set of equations for describing the free energy of a system based on parameters that adequately describes the chemistry and physics observed. While each program, model, and lab uses individualized sets of parameters, there are a few common to almost all MD simulations. Non-bonded interactions, such as Lennard-Jones, van der Waals, electrostatic, and chemical bonding interactions, are calculated for adjacent and spatially proximate atoms. Chain order gives insight into how tightly packed lipid tails are within the bilayer, which in turn has implications for the phase, melting temperature, and fluidity of the bilayer1. A completely saturated lipid tail, for example, would pack tightly and have restricted motion, but tails with unsaturated bonds would have kinks in the chain that cause it to pack less tightly3 (Figure 7.3.1).
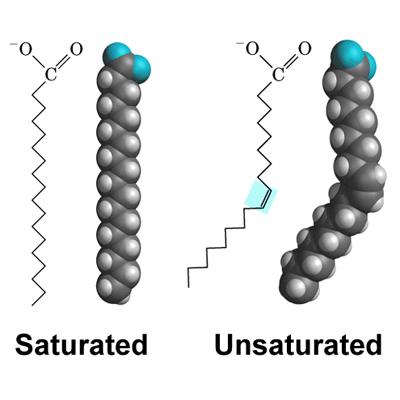
The size of the head group and lipid length further allow us to constrain MD simulations by providing estimations of the order and phase of the bilayer. Radial distribution functions describe the morphology and structure of the membrane, which when coupled with information about the density of the structure can provide a holistic spatial view of the bilayer1.
Water and solvents pose sizeable challenges in MD, as they comprise a majority of the atoms and interact with molecules in a multitude of ways, depending on temperature, pressure, and proximity to polar and hydrophobic groups. While many models represent water as a liquid, this model is insufficient in instances when extreme temperature changes lead to phase changes. The most widely used water model that accommodates these phase changes is the SPC model, which restricts water-molecule interactions to the oxygen and treats the hydrogen as fixed charges4,5 (Figure 7.3.2)6. More will be said about solvents in the Implicit versus Explicit Solvent Model section.

Calculating Charge of a Membrane
Within an MD simulation, the charge must be equal to zero. However, we know that biomembranes usually contain charged regions, as well as unique long-range interactions not commonly found in small molecule interactions. The integral of the potential (V(r) for long-ranged interactions is:
∫∞0V(r)r2dr=∞
where r is the radius of the space being modeled. For simplicity, this shape is assumed to be a symmetric sphere. That this integral diverges is problematic, but a number of techniques have been developed to address the charges indirectly. One solution (the Particle Mesh Ewald technique) applies different calculations that fall above and below a specified charge cut-off. Below the cut-off, electrostatic interactions are calculated directly, and above the cut-off they are calculated with Fourier analysis using a 3-D fast Fourier transform7. This method is preferred because it is highly accurate in the local environment while also reasonably fast. The above integral differs from the integral for Lennard-Jones and short-range interactions, which converge and can be easily calculated.
Force Fields
A force field is a set of parameters and equations that determine potential energy and therefore movement of a system.23 The four common and updated force fields for biological molecules are Amber, CHARMM, GROMOS, and OPLS. Each undergoes revision as experimental techniques change. Although each of the aforementioned force fields is commonly used for biomolecules, study of phosophilipids is most developed in the CHARMM and Berger force fields.
The CHARMM27 force field is an all-atom force field for lipids, nucleic acids, and amino acids that is being developed by various scientists including Martin Karplus. This force field is part of the CHARMM package of force fields. The Lennard-Jones potential is similar to other force fields but the electrostatic component is purely repulsive. This
The Berger force field is an addition to parameters found in Amber and OPLS that uses a united-atom force field that groups each carbon in a chain with its hydrogens. Compared to the CHARMM27 force field, this decreases the number of atoms in a system by one-third and drastically decreases the number of pairwise interactions. This method requires less computing power but is less accurate than all-atom force fields assuming that error is not large from step to step.
Membrane Protein Simulation
Proteins are an important part of cell membranes and should be considered when modeling biomembranes. The proteins along and inside membranes are the basis for many of the cell's functions including: signaling, motility, contraction, energy generation and consumption, and regulation. Use of MD to understand the behavior of membrane proteins in different situations is then an important technique in learning about the individual mechanisms of each protein.
Kandt et al. described a four step system to set up a membrane protein simulation23:
1) Orient the protein
To orient the protein, the protein and bilayer boxes should be the same dimension. Then the hydrophobic transmembrane domain is aligned with the lipid tail locations. This can be easily done with graphical interfaces and in GROMACS using the editconf tool.
2) Prepare the bilayer
There are multiple techniques, but the simplest and most common technique is to populate the lipid coordinates with lipids and then use cut-off lengths to delete physically impossible or improbably configurations. The problem is that local density requires additional simulation time to return to equilibrium, which can lead to larger error. In non-equilibrium simulations this is less of a problem, but the timescale required to achieve equilibrium may be in the nanosecond range, similar to a complete simulation.
A more efficient method is to create a lattice of lipids with a spacing greater than a real bilayer, insert the protein, and then compress the bilayer. The compression step goes through two repeating sub-steps: compression and energy minimization. This can be done with a grid and then compressing the lipid box size, or by superimposing the protein, expanding the bilayer to prevent overlap, and then scaling lipid positions.
3) Solvate the system
Adding water into bilayers is easy and quick, but proteins complicate this. Issues include water that may be trapped in the membrane by the protein and cavities in proteins that are isolated from the bulk. The simplest approach is to run a cavity analysis to avoid placing water in areas that they should not exist. In GROMACS, van der Waals radii are stored in vdwradii.dat and are used by the genbox tool. By increasing the vdW radii of lipid tails, no water will be present in the protein or bilayer.
4) Equilibrate the structure
Pressure coupling is an important part of achieving a realistic equilibrium. Isotropic pressure coupling does not allow for membrane fluctuations, while anisotropic coupling may cause systems to deform. Semi-isotropic pressure coupling should be used in interfacial systems, where x and y directions are coupled.
Electrostatics is another parameter, which is discussed by the section "Calculating Charge of a Membrane". In short, pairwise interactions are considered below a cut-off radius, and above the cut-off Fourier analysis is used to determine the interactions.
Then the simulation should be run to a minimum length to equilibrate the system. In example DPPC bilayers, it takes 10-20 ns of simulation to equilibrate. This is comparable to full simulation times so steps should be taken to optimize this step. Rotational and lateral diffusion is a rate limiting step, so the lipid bilayer should be set up such that this time is minimized. Protein coordinates must be restricted.
Potential energy will drop and minimize quickly but this is not a sign of equilibrium. Instead, look at pairwise interaction energies.
Odd structures
Other molecules are often associated with membrane proteins such as co-factors or ligands that may not exist in the simulation package23. Two solutions to this may be to use a force field with the required topology or to build it yourself. To re-build complex molecules, one common approach is to approximate the structure using amino acids if the structures are similar, or to find something else in the force field that can approximate it. This way the same force field and therefore set of parameters may be used for both the membrane protein and the lipids. Another consideration is that the membrane environment behaves differently compared to a water system. Using force fields meant to be in water causes problems with membrane protein structure and function, for example a potassium channel accepting sodium25.
Implicit versus Explicit Solvent Models
Implicit solvent models treat the solvent as a continuum26. The solvent is approximated as a field with an associated dielectric constant. Solvation free energy of a solute is incorporated as an effective energy term in the force field. This approach increases computational efficiency by eliminating solvent-solute interaction potentials. It also reduces degrees of freedom in the system, resulting in reproducibility between simulations and instantaneous solvent relaxation in membrane systems.
Explicit solvent models treat the solvent as individually bonded atoms. An all-atom model is used in these systems. For these, the choice of force fields and cut-off radius is a critical consideration. In the CHARMM27 force field, lipid tails trended towards all-trans compact conformations, which is smaller than reality. The CHARMM36 force field agreed with experiments much more. Also, orientation of membrane protein complexes is needed, or else solvents may be trapped or impossible bending situations may happen. Explicit solvents are much more computationally expensive.
Atomistic vs. Coarse-grained modeling
While atomistic models give us insight into the exact interactions happening within a membrane, this level of detail is computationally expensive. Modeling larger-scale processes such as bilayer assembly, lipid-protein interactions, and phase transitions with this method is not practical. In order to accommodate the amount of computing power these large scale simulations demand, coarse-grain models (CG) neglect details that do not affect long scale events. CG models minimize sampling by merging atoms into particle groups, or “pseudo-atoms,” based on shared chemical and electrostatic properties8. For example, a lipid might be condensed to a polar “head” molecule, a phosphate group, and a hydrophobic tail group (Figure 7.3.3). Another technique used to reduce degrees of freedom in a CG model is to ignore short-range dynamics. When looking at larger scale events, these interactions tend to have negligible effects, and are effectively unimportant for the question of interest9. Larger scale MD models, such as coarse-grain models, utilize, unsurprisingly, larger scale parameters. Two common parameters are the space occupied by individual molecules as well as membrane thickness. These properties are calculated using a variation on standard area and volume equations1.

Applications
Now that we have addressed some of the variables that go into constructing MD simulations of membranes, we will discuss some applications of this technique. This list merely glosses over an extensive field of research, but should provide an overview of the breadth of knowledge possible to acquire with these techniques.
We know that most membrane molecules engage in some degree of hydrogen bonding, but this is effectively impossible to observe experimentally. Atomistic models that utilize the expected geometry of a hydrogen bonded system create a functionally relevant picture of these interactions. Bond angles and lengths must be constrained to those observed experimentally, but accurate models have been generated this way10. In modeling hydrogen bonding, it is also crucial to constrain which functional groups can serve as bond donors and acceptors11.
MD simulations can be used to study phase transitions and formation of lipid rafts. By varying the temperature of the model, researchers can observe how classes of membrane molecules interact and either localize or disperse among other membrane molecules12,13. The rigidity as well as the fluidity of membranes can be measured using MD; these properties are determined by observing the density and lipid profile of the membrane. The way in which cholesterol contributes to both rigidity and fluidity in membranes has been studied extensively in membranes14.
Finally, protein-membrane and small molecule-membrane interactions two rapidly growing fields of research currently being pursued using MD15,16,17. While there are too many examples to include in this review, we will note that ion channels, cell surface receptors, and cytoskeleton proteins have all been studied with MD18,19.
Comparison with Monte Carlo Techniques
MD was created in response to the success of Monte Carlo techniques. In Monte Carlo (MC), random sampling is constrained by rules from probability, process rules, and statistical physics in order to generate a series of states27. These techniques are used for many applications from physics and chemistry to gambling. Current MC techniques are based on Markov chains, models that find the probability of the next event only based on the previous state. Random numbers are generated as start points with random variables and statistical ensembles constraining the system.
Compared to MD, MC is much less computationally expensive and is easier to implement. However, defining and viewing physical movement of systems is difficult and some trajectories may take particles into impossible positions. It does not show intermediate states as well as MD, and energy within the system is not so easily tracked or taken into account. For low density systems, the disadvantages of MC are not as important as molecules interact less often.
References
- 1Dickey AN and Faller R. Molecular Modeling of Biomembranes: A How-To Approach. In Biomedical Applications of Biophysics; Jue, T., Ed.; Humana Press, Springer, 2010; pp 35-58.
- 2Verlet L. 1967. Computer ‘experiments’ on classical fluids, I: thermodynamical properties of Lennard-Jones molecules. Phys Rev 159: 98-103.
- 3Tieleman DP, Marrink SJ, Berendsen HJC. 1997. A computer perspective of mebranes: molecular dynamics studies of lipid bilayer systems. Biochim Biophys Acta 1331: 235-270.
- 4Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. In Intermolecular forces; Pullman B.; Ed.; Dordrecht, Reidel, 1981; pp 331-342.
- 5Berendsen HJC, Grigera JR, Straatsma TP. 1987. The missing term in effective pair potentials. J Phys Chem 91: 6269-6271.
- 6Werten PJL, Rémigy HW, de Groot BL, Fotiadis D, Philippsen A, Stahlberg H, Grubmüller H, Engel A. 2002. Progress in the analysis of membrane protein structure and function. FEBS Lett 529: 65-72.
- 7Essman U, Perela L, Berkowitz ML, Darden HLT, Pedersen LG. 1995. A smooth particle mesh Ewald method. J Chem Phys 81: 3684-3690.
- 8Muller M, Katsov K, Schick M. 2006. Biological and synthetic membranes: what can be learned from a coarse-grained description? Phys Rep 434: 113-176.
- 9Lopez CF, Moore PB, Shelley JC, Shelley MY, Klein ML. 2002. Comoputer simulation studies of biomembranes using a coarse grain model. Comput Phys Commun 147:1-6.
- 10Lindahl E, Hess B, van der Spoel D. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model 7: 306-317.
- 11Dickey AN, Faller R. 2007. How alcohol chain-length and concentration modulate hydrogen bond formation in a lipid bilayer. Biophys J 92: 2366-2376.
- 12Jørgensen K, Mouritsen OG. 1995. Phase separation dynamics and lateral organization of two-component lipid membranes. Biophys J 69 (3): 942-954.
- 13Baoukina S, Mendez-Villuendas E, Bennett WFD, Tieleman DP. 2013. Computer simulations of the phase separation in model membranes. Farad Discuss 161, 63-75.
- 14Oldfield E, Chapman D. 1972. Dynamics of lipids in membranes: Heterogeneity and the role of cholesterol. FEBS Lett 23 (3) 285-297.
- 15Lee BW, Faller R, Sum AK, Vattulainen I, Patra M, Karttunen M. 2004. Structural effects of small molecules on phospholipid bilayers investigated by molecular simulations. Fluid Phase Equilibr 225: 63-68
- 16Lindahl E, Sansom MSP. 2008. Membrane proteins: molecular dynamics simulations. Curr Op Struct Biol 18 (4): 425-431.
- 17Bond PJ, Holyoake J, Ivetac A, Khalid S, Sansom MSP. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J Struct Biol 157 (3) 593-605.
- 18Isralewitz B, Gao M, Schulten K. 2001. Steered molecular dynamics and mechanical functions of proteins. Curr Op Struct Biol 11: 224-230.
- 19Paavilainen VO, Berling E, Falck S, Lappalainen. 2004. Regulation of cytoskeletal dynamics by actin-monomer-binding proteins. Trends Cell Biol 14 (7): 386-394.
- 20 Dill, K. A.; Bromberg, S.; Stigter, D. 2003, Molecular Driving Forces. New York: Garland Science.
- 21Berendsen HJC, Postma JPM, van Gunsteren WF, Dinola A, Haak JR. 1984. Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690.
- 22Posch HA, Hoover WG, Vesely FJ. 1986. Canonical dynamics of the Nose oscillator: Stability, order, and chaos. Phys. Rev. A. 33(6):4252-4256.
- 23Kandt C, Ash AL, Tieleman DP. 2006. Setting up and running molecular dynamics simulations of membrane proteins. Methods 41475-488.
- 24Sapay, N. and Tieleman, D. P. 2011. Combination of the CHARMM27 force field with united-atom lipid force fields. J. Comput. Chem., 32: 1400–1410.
- 25Domene, C., & Sansom, M. S. P. 2003. Potassium Channel, Ions, and Water: Simulation Studies Based on the High Resolution X-Ray Structure of KcsA. Biophysical Journal, 85(5), 2787–2800.
- 26Mori T., Miyashita N., Im W., Feig M., Sugita Y. 2016. Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochimica et Biophysica Acta, 1858:1635-1651
- 27Schlick, T. 2010. Molecular Modeling and Simulation: an Interdisciplinary Guide 2nd edition. New York: Springer.