
10.1: Hartree-Fock theory


- Last updated
- Oct 11, 2020
- Save as PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
The idea is to solve the Schrödinger equation for an electron moving in the potential of the nucleus and all the other electrons. We start with a guess for the trial electron charge density, solve Z/2 one-particle Schrödinger equations (initially identical) to obtain Z electron wavefunctions. Then we construct the potential for each wavefunction from that of the nucleus and that of all the other electrons, symmetrise it, and solve the Z/2 Schrödinger equations again.
Fock improved on Hartree’s method by using the properly antisymmetrised wavefunction (Slater determinant) instead of simple one-electron wavefunctions. Without this, the exchange interaction is missing. This method is ideal for a computer, because it is easily written as an algorithm.
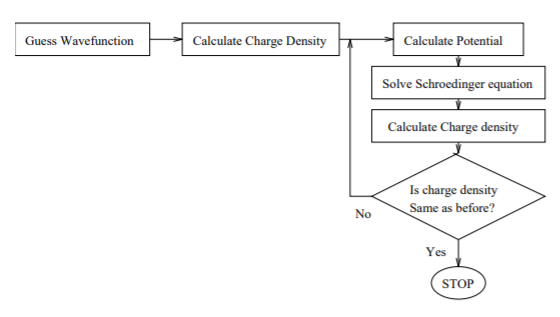
Figure 10.1.1 : Algorithm for Self-consistent field theory.
Although we are concerned here with atoms, the same methodology is used for molecules or even solids (with appropriate potential symmetries and boundary conditions). This is a variational method, so wherever we refer to wavefunctions, we assume that they are expanded in some appropriate basis set.
The full set of equations are
ϵiψi(r)=(−12∇2+Vion(r))ψi(r)+∑j∫dr′|ψj(r′)|2|r−r′|ψi(r)−∑jδσiσj∫dr′ψ∗j(r′)ψi(r′)|r−r′|ψj(r)
The first term is the kinetic energy and electron-ion potential. The second “Hartree” term, is the electrostatic potential from the charge distribution of N electrons, including an unphysical self-interaction of electrons when j=i. The third, “exchange” term, acts only on electrons with the same spin and comes from the Slater determinant form of the wavefunction.
Physically, the effect of exchange is for like-spin electrons to avoid each other. Each electron is surrounded by an “exchange hole”: there is one fewer like-spin electrons nearby than the mean-field would imply. The term i=j neatly cancels out the self interaction of the electron.