
X-ray Protein Crystallography
- Page ID
- 763
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)X-ray protein crystallography is a technique by which it is possible to determine the three dimensional positions of each atom in a protein. Now over 100 years old, x-ray crystallography was first used to determine the three dimensional structures of inorganic materials, then small organic molecules, and finally macromolecules like DNA and proteins. To date, about 100,000 protein structures have been published in the Protein Data Bank, with almost 10,000 added every year. To use this technique, the crystallographer obtains protein crystals, records the diffraction pattern formed by x-rays passed through the crystals, and then interprets the data using a computer. The result is a atomic-resolution model of a protein.
History
Though crystal symmetry was explored in the late 1600s by Danish scientist Nicolas Steno, and continuing efforts by René Just Haüy and William Hallowes Miller in 1839 firmly established that a crystal is a ordered lattice, it wasn't until the discovery of x-rays in 1895 and the proof of their diffraction by Max von Laue in 1912 that crystallography as a science began.
After the use of x-ray crystallography to deduce the lattice structure of table salt in 1914, the father and son team of William Henry Bragg and William Lawrence Bragg shared the 1915 Nobel Prize in Physics for the development of Bragg's law,
\[ n \lambda = 2 d \sin(\theta), \]
which relates an x-ray diffraction pattern with the three-dimensional structure of a crystal.
The field has received numerous Nobel Prizes over the years, including in chemistry in 1964 to Dorothy Crowfoot Hodgkin, who solved the structures of the small molecules cholesterol, penicillin, and vitamin B12, and in chemistry in 1962 to Max Perutz and John Cowdery Kendrew for their work on sperm whale myoglobin. David Chilton Phillips solved the first structure of an enzyme, lysozyme, in 1965.
The early 70s saw the birth of the Research Collaboratory for Structural Bioinformatics' Protein Data Bank. The PDB began with 13 structures in 1976 and has grown to the "single worldwide archive of structural data of biological macromolecules".
Technique
Obtaining crystals
The first and least certain step in crystallography of a protein is obtaining crystals of the protein of interest. Obtaining suitable amounts of the protein of interest is usually carried out in a straightforward manner using established molecular biology techniques such as molecular cloning and affinity chromatography. However, the crystallization step remains the bottleneck for this technique, with some proteins (particularly proteins that exist in the aliphatic environment of the plasma membrane) remaining intransigent to crystallization even in the face of the most diligent crystallographers. Thus, for each protein of interest, a large number of crystallization conditions must be tried, necessitating a relatively large amount (milligrams) of the pure protein.
Protein production and purification

To produce suitable amounts of protein, contemporary crystallographers turn to molecular biology's old friend Escherichia coli. A gene which codes for the protein of interest is cloned into a small, circular piece of DNA known as an expression plasmid. The expression of the gene is typically under the control of an inducible promoter, and is regulated by the researcher rather than the bacteria. Cells are transformed with the expression plasmid, grown to high density, and induced to express the protein of interest. The cells are lysed chemically with detergents or physically with sonication, and the protein is purified, typically via affinity chromatography. High purity (greater than 95%) is desirable. Often, it takes multiple experiments before the method that obtains maximum protein is found.
Crystallization
The concentrated protein solution obtained is then subjected to a wide variety of crystallization conditions. Since we have no way of knowing a priori which set of conditions is right for obtaining crystals of a given protein, many different conditions are tried in parallel using a technique called drop diffusion.
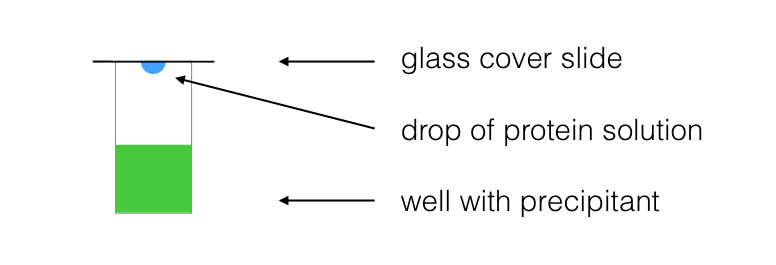
In this technique, a small quantity (typically a microliter) of concentrated protein solution is mixed with an equal volume of precipitant. This drop is separated by air from a large volume of precipitant solution. The drop is hypotonic to the precipitant and slowly equilibrates to the concentration of the large volume of precipitant. Concomitantly, the concentration of protein increases. If this process occurs at just the right rate, the protein precipitates out of solution into an ordered lattice structure: a protein crystal.
It is often said that this part of crystallography is more of an art than a science, and indeed there is little theoretical guidance available to the crystallographer who wishes to crystallize a new protein. Patience, and to some extent, luck, determine the sucess or failure of the crystallization of any particular protein.
Obtaining x-ray diffraction data
Once crystals of suitable size and composition are obtained, it is necessary to bombard the crystal with x-rays and observe the diffraction pattern. An x-ray diffractometer works in a similar manner to a light microscope. In a light microscope, the subject is irradiated with visible light (400 nm < \( \lambda \) < 700 nm$), which is diffracted by a lens onto the retina, producing a macroscopic image of a microscopic object. Molecules such as proteins are much smaller than microscopic structures like cells, and, as such, require that a shorter wavelength of radiation be used during diffraction. X-rays, where $100 pm < \(\lambda\) < 10,000 pm$, are the perfect size to diffract around atoms (32--225 pm), bonds (74--267 pm), and molecules (100 pm to hundreds of Angstroms). However, x-rays are difficult to focus in a manner analogous to the way a lens focuses visible light. Crystallographers employ computational methods to capture the x-ray scattering pattern (pictured at right) and infer the three-dimensional positions of atoms in a molecule.
X-ray sources
Traditionally, x-ray crystallographers filtered and directed the x-rays generated by radioactive cesium in their diffractometers, but today it is much more common to use synchrotron radiation to irradiate samples. Synchrotrons, huge hollow rings used to accelerate electrons for use in studies of subatomic particles, produce huge amounts of tunable (different wavelengths) x-ray radiation that is perfect for irradiating crystals.

Sample preparation
The crystal is suspended in aqueous solution containing a cryoprotectant in the eye of a small loop. The crystal and loop are cooled with a continuous stream of liquid nitrogen to prevent chemical damage by the x-rays. X-rays are directed through the crystal, and the diffraction pattern at any given moment is recorded by a detector. The crystal is rotated sightly and a new diffraction pattern is obtained. This process is repeated through 360 degrees along one axis (typically rotations through a smaller angle on another axis are also recorded to avoid blind spots) until the instrument has recorded a diffraction pattern for each position.
X-ray scattering
As an incident x-ray (electromagnetic wave) overlaps with an electron, it is elastically scattered, generating a secondary wave that has the same wavelength, but different direction, than the incident wave (thus the wave is "scattered" or "diffracted"). Due to the symmetry of the crystal and its many repeated units, these secondary waves interfere constructively at only one point along a circle drawn around the atom that scattered them. It is that point, described by Bragg's Law, that appears as a dark spot on the detector. An example diffraction pattern, from a SARS protease, is displayed at right.
Obtaining an electron density map
The data recorded by the detector during diffraction are now subjected to computational analysis. First, each spot in each diffraction image is indexed, integrated, merged, and scaled by a computer, producing a single text file from thousands of images. The position of each spot depends on the properties of the crystal, and as such is different for every protein. The process of converting the reciprocal space-representation of the crystal into an interpretable electron density map is known as phasing.
Shown below is the software PyMOL displaying the electron density map (white) for Protein Data Bank structure 4BLL, a peroxidase from the model organism Pleurotus ostreatus, overlayed with the model from the PDB structure (pink).

Fundamental challenge of crystallography
The problem is this: the detector is only able to record the position and intensity of an x-ray when it hits the detector. An x-ray has both intensity, which is related to the amplitude of the wave, and phase, which is related to the point at which the x-ray was scattered. The crystallographer would dearly like to know the phase of an x-ray, because this information along with with intensity, but the detector is not capable of capturing phase information due the quantum mechanical nature of x-rays and electrons.
Overcoming the challenge: phasing
Crystallographers use several methods for recovering phase information from diffraction data. Common techniques include:
- direct methods
- molecular replacement
- anomalous x-ray scattering
- multiple isomorphous replacement
Direct methods use the Sayre equation to determine phases directly from the diffraction data. These methods are only viable for small (less than 1000 atoms) molecules and are not typically used in protein crystallography. The 1985 Nobel Prize in chemistry was awarded to Hauptman and Karle, who developed these methods.
The technique of molecular replacement uses the solved crystal structure of a homologous protein to provide a "seed" electron density map that can then be refined by a computer. Molecular replacement is used extensively in labs that solve the crystal structures of several mutants of a given protein.
Anomalous x-ray scattering relies on protein production in a host that is incapable of producing the amino acid methionine. The host instead uses a synthetic amino acid, selenomethionine, in which methionene's sulfur atom has been replaced by selenium. The positions of any selenomethionines in the diffraction pattern can be solved using different x-ray wavelengths and direct methods, and the rest of the structure can be solved using the position of the selenomethionines as a reference.
Multiple isomorphous replacement has largely been superseded by anomalous x-ray scattering and works in a similar way, except using metal ions instead of synthetic amino acids for initial phasing.
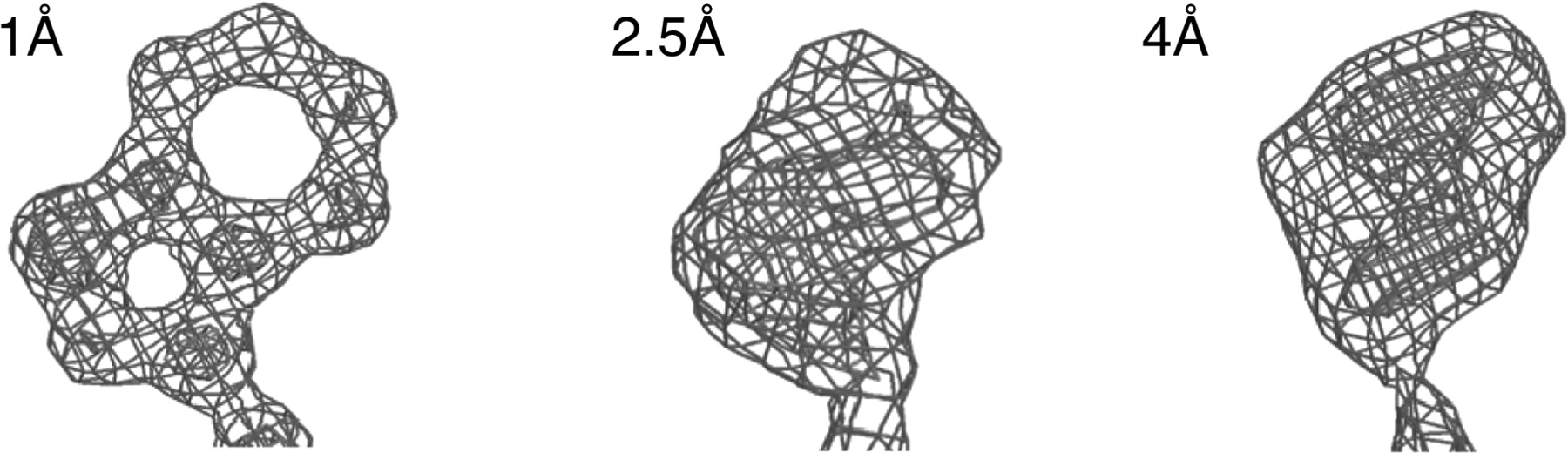
Once phases have been recovered, it is possible to mathematically reconstruct the positions of electrons within the crystal using Fourier synthesis. (The diffraction data is the Fourier transform of the electron density in the unit cell.) A computer applying these operations with correct phases constructs a three-dimensional map of electron density map than can be viewed with molecular visualization software. The resolution of the data determines the resolution of the model, as depicted below with electron density maps of tryptophan at three different resolutions.
Obtaining a three-dimensional model
With sufficient resolution (less than 1.5 A), it is possible to automatically generate a model based on the electron density map and known bond angles and lengths, and known sizes of atoms. In practice, not all crystallography data is of such high quality. Often, the crystallographer uses molecular visualization software to manually fit a chemical model to the electron density data. The result is a model that can be viewed with molecular visualization software. An example, draw in PyMOL from PDB structure 4BLL, is below.

Further reading
- Bragg WL (1914). "The analysis of crystals by the X-ray spectrometer". Proc. R. Soc. Lond. A89 (613): 468
- Glusker JP and Trueblood KN, 1972. Crystal structure analysis: a primer. Oxford University Press. [Reprint: OUP Oxford, May 27, 2010]
- Drenth J. Principles of Protein X-Ray Crystallography. Springer, Apr 5, 2007
- Hauptman H, 1997. "Phasing methods for protein crystallography". Curr. Opin. Struct. Biol. 7 (5): 672–80
- Chernov AA (2003). "Protein crystals and their growth". J. Struct. Biol. 142 (1): 3–21